
Abstract
Tumor necrosis factor (TNF)-α is a potent trimeric cytokine which plays a fundamental role in the host immuno-inflammatory response, as well as in homeostasis and development. Although critical for canonical immune function, TNF-α has great destructive potential and is implicated in the development of multiple immune-mediated disorders. Within the context of rheumatoid arthritis (RA), TNF-α acts as a primary pathogenic driver by precipitating a pro-inflammatory cytokine cascade and coordinating the attraction and activation of immune cells, all of which culminate in damage to the synovium. The discovery of the paramount role of TNF-α in the pathophysiology of RA motivated studies to understand the effects of TNF blockade in vitro and in vivo. Promising preclinical results provided the impetus for clinical trials, spearheaded in the 1980s and 90s by Marc Feldmann and Ravinder N. Maini, which revealed significant improvements across RA symptom scores and finally led to approval by the Food and Drug Administration (FDA) in 1998. As of 2021, five TNF-α blocking agents have been widely applied clinically, including infliximab (IFX), etanercept (ETN), adalimumab (ADA), golimumab (GLM) and certolizumab pegol (CZP). All of them successfully ameliorated symptoms of RA and the associated tissue damage, especially in patients not responding to traditional treatment methods. Anti-TNFs are most often administered in combination with methotrexate (MTX) as part of Phase II treatment (i.e., second line). Although the general availability of anti-TNFs has dramatically improved patient outcomes, sustained remission is rare, and the mechanism of RA remains incompletely understood. Thus, additional basic and translational research with the aim to develop novel RA treatments is warranted.
Introduction
Rheumatoid arthritis (RA) is a heterogeneous autoimmune disease characterized by chronic and systemic inflammation, leading to progressive deterioration of joints 1. Symptoms of RA severely impact the quality of life of patients and can lead to marked functional impairment and disability. Traditional treatment regimens include glucocorticoids (GCs), nonsteroidal anti-inflammatory drugs (NSAIDs), and disease modifying antirheumatic drugs (DMARDs) 2. However, these treatment regimens are often insufficient to produce sustainable limitations of disease progression and may even lead to disease exacerbation 3. Since the 1980s, continuous progress has been made in establishing tumor necrotic factor-α (TNF-α) as a critical mediator of the pathogenesis and progression of RA 4,5.
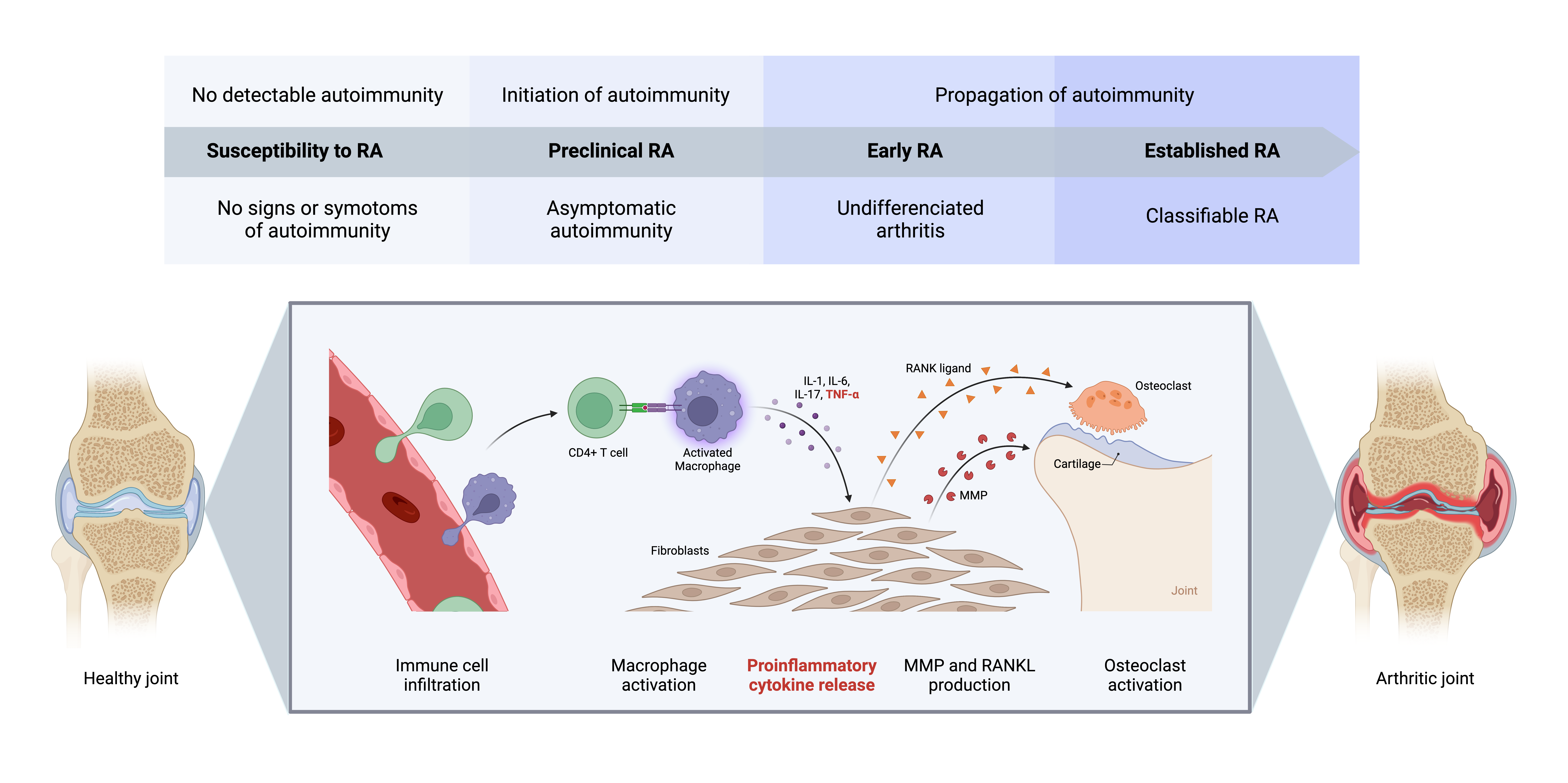
Figure 1. Pathogenesis and disease progression of rheumatoid arthritis. In a susceptible joint (knee joint illustrated herein), a not yet known trigger promotes inflammation in the synovium, initiating the infiltration of immune cells including, but not limited to, CD4+ T cells and macrophages. Autoreactive CD4+ T cells then activate macrophages, which produce a milieu of proinflammatory cytokines, including TNF-α. Stimulated by the foregoing cytokines, fibroblasts produce MMP, which causes direct tissue destruction, and RANKL, which activates osteoclasts. Together, the foregoing factors cause joint destruction. Graphics created with BioRender.com.
Role of TNF-α in Immunity and Homeostasis
Tumor necrosis factor (TNF) was named after its discovery process in the 1890s wherein Dr. Williams B. Coley, a pioneer of modern immunotherapy, successfully induced tumor necrosis by injecting inoperable sarcoma patients with crude bacterial extracts termed “Coley’s Toxin” 6, 7. Although his treatment was largely successful, patients curiously developed systemic hyperinflammation 7. TNF, the molecule responsible for in vitro lysis, was later isolated from the serum of endotoxin-treated animals 7. In 1985, Nobel Prize winner Dr. Bruce Beutler purified TNF-α, originally named ‘cachectin’ due to its ability to induce cachexia, from the supernatant of endotoxin-treated macrophages 8.
TNF-α, a member of the TNF superfamily, is a conserved trimeric cytokine with diverse systemic and local effects 7. Although largely produced intracellularly by macrophages and dendritic cells, TNF-α can affect most immune and non-immune cells 9. TNF-α release is classically induced by lipopolysaccharide (LPS), however many other in vivo and in vitro stimulators of TNF-α are known 10. Two forms exist: the membrane-bound (mTNF-α) and soluble form (sTNF-α) 7. The expression of TNF-α is NF-κB-dependent, as evidenced by multiple NF-κB binding sites present within the promoter region of the TNF gene 7. TNF-α exerts its molecular signalling functions, including the induction of tumor necrosis, via its interactions with TNF receptors (TNFRs), TNFR1 (expressed by a wide variety of cells) and TNFR2 (expressed mostly on lymphocytes, both of which are found in membrane-bound or soluble (e.g., p75) forms, instigating homodimerization and intracellular signaling cascades 7.
The two classes of TNF-α receptors produce antagonistic effects: TNFR1 (death receptor with death domain) signaling induces inflammation, host defense, and apoptosis or necroptosis, whereas TNFR2 is anti-inflammatory and pro-cell proliferation 9. When TNFRs were knocked out in transgenic mice, they became more susceptible to infections and displayed a suppressed inflammatory response, as measured by a decreased response to LPS challenge 11, 12. Disparate actions are mediated by the specific recruitment of adaptor proteins (e.g., TNFR-associated factors: TRAFs), activation of kinases, and phosphorylation/dephosphorylation events 7. Notably, TNF-α autocrine and paracrine induction of cytokines and/or cell survival factors is dependent upon NF-κB translocation to the nucleus 7.
The three main events mediated by TNF-α signaling are: acute and/or chronic inflammation (notably host response to infection), apoptosis, and necroptosis 7. TNF-α plays a fundamental role in the inflammatory and immune responses by recruiting immune cells to sites of infection, stimulating macrophage phagocytosis, as well as regulating and acting in concert with other pro-inflammatory cytokines 13. Although crucial for the local containment of infection, systemic release of TNF-α can induce septic shock 14. Homeostatic roles include, but are not limited to, effects on bone remodeling, and sleep-wake cycles, regulation of embryonic development and cell differentiation, as well as lymph node follicle and germinal center formation 7, 15. Taken together, TNF-α is a potent pro-inflammatory cytokine implicated in multifaceted pathways across the immuno-inflammatory axis, in homeostasis, and in development, thereby demonstrating both destructive and protective potential.
Role of TNF-α in Rheumatoid Arthiritis
One of the hallmarks of RA is an inflamed synovial membrane that continuously releases pro-inflammatory cytokines, leading to the destruction of bone and cartilage in the joints 2,16. Due to the potency of TNF-α as a pro-inflammatory cytokine, there have been continual efforts since the 1980s to investigate its role in inflammatory diseases such as RA12, 17. Researchers have gathered the following evidence to verify the involvement of TNF-α as a mediator in the pathogenesis of RA: (1) TNF-α is one of the most abundantly produced cytokines at the site of the inflamed synovial membrane; (2) the presence of high levels of TNF-α has been associated with active tissue damage; (3) both in vitro and in vivo experiments have demonstrated that TNF-α can induce damage to normal cartilage; (4) the induced tissue damage can be prevented by antagonization of TNF-α using neutralizing antibodies 4,18. As such, it is well-supported that TNF-α plays a crucial role in the propagation of inflammation and the destruction of tissues in RA.
The pathophysiology of RA is subserved by a complex genetic architecture coupled with environmental factors 19. Notwithstanding, convergent lines of evidence demonstrate that synovitis (inflammation of the synovial membrane), a hallmark of RA, is caused by the infiltration and local activation of mononuclear cells in the synovial membrane 20. In susceptible individuals, it is surmised that the foregoing process is triggered by the activation of innate immunity (primarily through TLR signaling), which enables antigen-presenting cells to display arthritis-associated antigens to T cells 20, 21. Consequently, CD4+ T cells infiltrate the synovium whereupon they attract and coordinate the activation of lymphocytes, monocytes, macrophages, and fibroblast-like synoviocytes, among other events 20.
The aforementioned cells, in concert with macrophages activated directly through TLR signaling, produce pro-inflammatory cytokines including TNF-α, which can be detected in the synovial fluid of RA patients 20. A critical node in the etiology of RA, TNF-α plays both systemic and local roles, and many of its effects have been reasoned in the reverse through studying blockade. Notably, TNF-α is a key regulator of the inflammatory cascade, inducing the production of a proinflammatory mosaic consisting of cytokines such as interleukin (IL)-1, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF), which jointly orchestrate cellular responses resulting in structural and functional damage to the joint 14, 20, 21. TNF-α is also thought to stimulate mesenchymal cells to release matrix metalloproteinases (MMPs), which destroy tissue, and to bolster MMP effects through the concomitant inhibition of MMP inhibitor production 20. As a corollary, through the stimulation of IL-11, TNF-α indirectly promotes the development of osteoclasts, as well as drives osteoclastogenesis by stimulating the expression of receptor activator of nuclear factor kappa-Β ligand (RANKL) and M-CSF 20, 21. TNF-α also precipitates bone resorption through the upregulation of prostaglandins and leukotrienes, potentiates angiogenesis through the upregulation of vascular endothelial growth factor (VEGF), as well as induces the proliferation of fibroblasts and expression of adhesion molecules to facilitate trafficking of leukocytes to the joint 20, 22.
Taken together, the confluence of proinflammatory and physiological actions induced by TNF-α precipitates articular cartilage damage facilitated by osteoclasts, chondrocytes, and synovial fibroblasts, enabling synovial hyperplasia and formation of the pannus 21. TNF-α overproduction in arthritic joints can also promote increased circulating levels of TNF-α, driving cardiovascular disease through insulin resistance (due to tissue alterations), free fatty-acid release from adipocytes, and upregulation of pro-coagulant proteins, as well as induction of acute-phase protein production, and HPA axis dysregulation 21, 22.
History of TNF-α blocking treatments in RA
Pre-clinical models
Prior to the advent of biologic agents for RA, treatment options consisted of repurposed or serendipitously discovered pharmacotherapies. The foregoing options produced unsatisfactory outcomes in a substantial patient subset (non-responders). As such, the development of novel treatments with the aim of improving patient outcomes was a key research focus.
The first cytokine to be implicated in the pathogenesis of RA was IL-1, detected in the synovial fluid of RA patients in 1982 22. Subsequently, through the utilization of cytokine assays of RA synovium culture supernatants, other cytokines including TNF-α, IL-6, and GM-CSF were discovered to be continuously produced in the diseased joint 23. However, due to their redundancy of action, it remained unclear which cytokine was paramount and could thus serve as a therapeutic target 24. The observation that TNF-α was the first cytokine produced by macrophages during an inflammatory response was one of the first clues towards its critical importance 25. Furthermore, a key experiment by Fiona Brennen and Marc Feldmann demonstrated that blocking the action of TNF-α in culture inhibited the production of IL-1 (and IL1B mRNA), another prominent cytokine thought to be implicated in RA due to known in vitro joint-damaging actions 26-28. To parse out the critical role of TNF-α at the apex of the pro-inflammatory cascade, experiments were conducted to demonstrate the efficacy of anti-TNF-α in downregulating the production of multiple inflammatory cytokines 29. The foregoing results strengthened the hypothesis implicating TNF-α as a key driver of RA pathogenesis, thus, investigators became optimistic that blocking TNF-α in vivo would ameliorate RA symptoms.
Marc Feldmann and Ravinder N. Maini conducted a pivotal preclinical investigation wherein anti-TNF-α antibodies were administered to genetically susceptible mice with collagen-induced arthritis (a model of RA). Results were encouraging; treatment before or after the onset of disease reduced paw swelling and other clinical indicators of disease progression 30. Other murine models of RA also responded positively to high-dose treatments, whereas exogenous administration of TNF-α worsened the disease course 30. Moreover, transgenic overexpression of the human TNF-α gene produced an RA-like phenotype in mice 31, 32. Taken together with in vitro experiments, the foregoing results were the impetus to test TNF-α blockade in human RA patients; efforts facilitated by the pre-existence of human anti-TNF-α, which had been unsuccessfully developed for the treatment of septic shock 33.
Clinical trials
In 1992, Marc Feldmann and Ravinder N. Maini (with involvement of pharmaceutical company Centocor through Jim Woody) led a Phase I/II clinical trial to test the efficacy of anti-TNF-α antibodies in human RA patients at Charing Cross Hospital 34. The American College of Rheumatology criteria is most commonly used to assess the efficacy of RA treatments in clinical trials. Efficacy is operationalized as a composite 20% reduction of clinical and functional measurements (ACR20), including number of tender and number of swollen joints, functional ability measure, and CRP levels 35. Results of the 1992 trial were promising; 20 RA patients refractory to multiple treatments exhibited tolerability as well as rapid improvement in clinical symptoms (by ACR criteria) and decreases in serum pro-inflammatory cytokines 34. Although patients relapsed following discontinuation of treatment and the open study design was vulnerable to confounding by the placebo effect, promising preliminary results supported further investigation 34.
As such, in 1993 Marc Feldman and James Woody initiated a Phase II, randomized, double-blind placebo-controlled trial at 4 European centers. Treatment was well-tolerated, and the trial demonstrated formal proof of efficacy; by the endpoint at 4 weeks, 79% of patients had responded to 10 mg kg−1 (high dose), 44% to 1 mg kg−1 (low dose), compared to an 8% response to placebo 3 6. However, questions remained regarding long-term efficacy and tolerability as even fully humanized antibodies have the potential to produce immunogenicity 24, 37. To address such concerns, Marc Feldmann and collaborators conducted a long-term Phase II 26-week, double-blind, placebo-controlled study evaluating the efficacy of anti-TNF-α alone or in combination with methotrexate in 101 RA patients. Results revealed that approximately 60% of patients achieved clinical improvement, operationalized as the 20% Paulus criteria for response to treatment 38. Low doses were effective at the start of treatment, however, only intermediate (3 mg kg−1) and high doses produced long-lasting effects. Furthermore, Feldmann’s 26-week RCT demonstrated that immunogenicity was tolerable, inversely related to dose, and reduced by co-treatment with methotrexate (MTX) 38.
The addition of MTX as a co-therapy produced improved outcomes in a Phase III trial lasting 2 years (termed ATTRACT) evaluating treatment of anti-TNF-α (infliximab) for the reduction of RA symptoms and radiographic progression (cartilage damage and bone destruction) 39. Moreover, patients receiving combination therapy reported improved quality of life, and approximately half demonstrated repairs in joint damage 40. Clinical improvements in RA symptoms using anti-TNF-α were mediated by abrogation of many of the pathophysiological actions of TNF-α; reduction in the levels of pro-inflammatory cytokines, leukocyte trafficking, angiogenesis, joint destruction, decreased cardiovascular disease risk through hematological normalization, and overall normalization of the immune response 39. Although anti-TNFs served to remove the linchpin of RA progression, they could not reverse existent damage. Notwithstanding, combination treatment consisting of an anti-TNF-α with MTX was approved for use in the United States and Europe in 1998 24. Subsequently, multiple clinical trials were conducted to demonstrate the efficacy of other proprietary anti-TNFs 39.
The three leading TNF-α blockade candidates for clinical trials were Infliximab (INF), Etanercept (ETA) and Adalimumab (ADA). The following section aims to discuss in further detail the efficacy and safety of INF, ETA and ADA, in addition to two newer TNF-α blocking agents, golimumab (GLM) and certolizumab pegol (CZP).
Currently used TNF-α blocking agents
Infliximab (INF)
INF was the first TNF-α blocking agent to be clinically tested in RA patients. It is a chimeric monoclonal antibody with its constant region, or fragment crystallization (Fc) region, derived from human IgG1κ and its variable region, or fragment antigen binding (Fab) regions, composed of the Fab regions of a high-affinity neutralizing anti-TNF-α antibody of murine origin 41-43. INF not only prohibits the binding of TNF-α to TNFRs, but also promotes the dissociation of TNF-α molecules from TNFRs 44. INF has a half-life of 8-10 days and needs to be administered intravenously every 4-8 weeks 41, 42.
Several randomized, placebo-controlled clinical trials tested the safety and efficacy of INF in treating RA patients 45-48. The doses they tested ranged from 3-10 mg/kg. Significant improvements were seen in patients treated with a combination of INF and MTX as compared to MTX plus placebo as assessed by ACR criteria 45-47. The amelioration of joint destruction was long-lasting, as evident upon radiographing 45. Interestingly, there was no significant correlation between clinical outcomes and the dosage of INF treatments 45-48. The major side effects associated with INF treatment include common infusion reactions (e.g., injection site pain) and increased risk of infection 4,41,49, 50. The rates of serious infection and life-threatening infusion reactions, however, were similar in groups treated with INF plus MTX and placebo plus MTX 4. There also exists the possibility that the risk of malignancies and of cardiovascular conditions is elevated among the INF-treated group 50.
Etanercept (ETA)
ETA is a dimeric fusion protein with the Fc domain of IgG1 linked to the extracellular portion of the TNFR2. Since ETA has a higher affinity for TNF-α than natural TNFRs, it inhibits the activity of TNF-α signalling pathways by salvaging soluble TNF-α. ETA is a fully human protein, so the risk that it will induce neutralizing antibodies is lower than the risk that chimeric proteins will. ETA has a half-life of 3-5.5 days and is administered subcutaneously every 1-2 weeks 51.
Several randomized, placebo-controlled clinical trials tested the safety and efficacy of ETA in treating RA patients 52–55. The recommended dose for ETA is 50 mg/kg when administered once a week 66. Patients treated with ETA showed significant improvements when compared to placebo controls as assessed by the ACR criteria 4,53,51. Furthermore, treating with a combination of ETA with MTX resulted in greater improvements than treating with ETA or MTX alone did 8. Increasing the dose to 50 mg/kg twice a week, did not yield significantly better results than the recommended dose 56. The major side effects of ETA include infusion reactions and hypertension, although the symptoms are often mild to moderate. No difference in the rate of severe adverse effects, including infections, was observed between the ETA-treated group and the control group 41,57.
Adalimumab (ADA)
ADA is a high-affinity and fully human monoclonal antibody for TNF-α molecules 58, 59. This recombinant monoclonal antibody is of class IgG1. Its mechanism of inhibiting the activities of TNF-α is similar to that of INF (i.e., prevention of receptor binding). Since ADA is structurally indistinguishable from natural human IgG1 molecules, there is minimal risk of decreased half-life attributed to the induction of neutralizing antibodies 60. ADA has a half-life of 10-20 days and the FDA recommends intravenous or subcutaneous injections twice a week 41.
Several randomized, placebo-controlled clinical trials tested the safety and efficacy of ADA in treating RA patients 61-64. The doses ranged from 0.5-10 mg/kg for intravenous injections or 10-80 mg/kg for subcutaneous injections. Greater improvements were observed in patients treated with ADA as compared to placebo controls, according to the ACR criteria. As with INF, ADA treatments ameliorated the progression of radiographic joint damage 63. Moreover, regimens combining ADA and MTX treatments showed superior clinical and functional improvements as compared to those treated with ADA or MTX alone 65. Overall, severe adverse effects associated with ADA were rare, signifying a high level of tolerance. Common side effects of ADA include infusion reactions and increased risk of infections, with the most elevated risk of serious infections at the 40 mg/kg dose 41,63.
Golimumab (GLM)
GLM is another fully human monoclonal antibody targeting TNF-α of the IgG1 subclass. It was one of the newer TNF-α blocking agents and was approved by the FDA in 2009 66. GLM is generated in an in vivo system where genetically modified mice are immunized with human TNF-α molecules. The mechanisms behind the TNF-α neutralizing actions of GLM are similar to that of INF and ADA, principally by promoting dissociation between TNF-α and TNFRs 67,68. GLM has a half-life of approximately 12 days and the FDA recommends monthly subcutaneous administrations 68.
Several randomized, placebo-controlled clinical trials tested the safety and efficacy of GLM in treating RA patients 69,70,67. The clinical trials tested two doses, 50 mg and 100 mg, with no statistically significant difference between the two in terms of clinical efficacy 41,66. According to the ACR criteria, patients treated with GLM plus MTX exhibited reduced RA symptoms and decreased risk of radiographic progression as compared to those treated with placebo plus MTX41,67. The most commonly reported adverse effects of GLM treatment include infusion reactions, hypertension, and increased risk of infections 68. The adverse effects were more prominent at the 100 mg dose, prompting the recommended dose to be set at 50 mg 66. These side effects are rare and the severity generally ranges from mild to moderate, demonstrating that GLM is generally well-tolerated 41.
Certolizumab pegol (CZP)
CZP is another newly developed TNF-α blocking agent and was approved for clinical use by the FDA in 2009. Unlike INF, ADA, and GLM, CZP contains only a single humanized Fab fragment with a high affinity for TNF-α, which is artificially conjugated to polyethylene glycol (PEG)71. Due to its unique structure, its mechanism of action is different from the other aforementioned TNF-α blocking agents 72,73. Specifically, due to the lack of an Fc region, CZP does not induce Fc-mediated cytotoxic effects, including complement-dependent cytotoxicity and antibody dependent cell-mediated cytotoxicity 71. As well, as compared to other TNF-α blocking agents, CZP has a longer half-life (around 2 weeks) and improved bioavailability 71,72. The FDA recommends CZP to be administered subcutaneously twice at 200 mg (for a total of 400 mg) initially at weeks 2 and 4, respectively, followed by a single 200 mg injection every two weeks.
Several randomized, placebo-controlled clinical trials tested the safety and efficacy of CZP in treating RA patients 74-77. Similar to other TNF-α blocking agents, when combined with MTX, GLM treatments led to significantly better clinical outcomes as compared to the placebo plus MTX control61. In addition, patients who underwent GLM monotherapy also showed rapid reductions in RA symptoms and slowed radiographic progression as compared to baseline 73, 77. Increasing the dose from 200 mg to 400 mg did not provide a higher level of clinical improvements, however, the severity of adverse events was higher in the 400 mg dose group 41,77. The most prominent adverse effect of CZP is an increased risk for tuberculosis. Other side effects include pain in the injection area and hypertension. These adverse events were mostly mild or moderate 76.

Table 1. Summary of half-life, dose and administration route, therapeutic outcomes, side effects, and structure of anti-TNFα agents. Dosing recommendations, therapeutic outcomes, and side effects have been extracted from the clinical trials that led to the approval of these agents by the FDA and Health Canada.
Risk and Adverse Effects of Anti-TNFs
Although anti-TNF biologics have had a dramatic impact on the quality of life of many RA non-responders, utilization is not without risk. Of critical importance is the consideration that blocking the effects of TNF-α dampens general immunity. As such, RA patients treated with anti-TNF biologics are at an increased risk of serious viral, bacterial, and fungal infections – notably opportunistic pathogens and intracellular bacteria 78. Furthermore, there is an increased risk of lymphoma and non-melanoma skin cancers, as well as reactivation of latent tuberculosis due to the breakdown of granuloma 79. Rarely, patients undergoing anti-TNF treatment may develop new or worsening demyelinating diseases and/or drug-induced lupus 80. Common side effects of anti-TNF biologics, many of which are injection-site reactions, include headaches, rashes, anemia, upper respiratory tract infections, cough, diarrhea, nausea, and abdominal pain 80.
Comparing TNF-α blockade to other rheumatoid arthritis therapies
Glucocorticoids and nonsteroidal anti-inflammatory drugs
Despite the potential for such adverse effects, TNF-α blockade is a striking improvement in the management of RA. In addition to lifestyle and physical approaches (e.g., exercise, occupational therapy), previous pharmacotherapies were largely diverted from other conditions. Willow bark derivatives (i.e., salicylates) have been used to provide symptomatic relief since antiquity. Furthermore, GCs and NSAIDs can function rapidly to reduce pain and swelling, so they are often prescribed shortly after the onset of clinical symptoms 81 NSAIDs, salicylate derivatives which inhibit cyclooxygenase (COX)-1 and 2 in order to reduce the production of prostaglandins, emerged in the mid-20th century, and derive their name by means of comparison to glucocorticoids (e.g., steroids such as cortisone), which were concurrently shown to reduce joint inflammation 82. With the progression of RA symptoms, however, GCs and NSAIDs often lose effectiveness, creating an unmet demand for more effective treatments, such as DMARDs and anti-TNFα blockade. Moreover, chronic use of NSAIDs may lead to gastrointestinal abnormalities (e.g., bleeding, nausea, ulcers), tinnitus and hearing loss.
Disease-modifying antirheumatic drugs
Drugs which slow the course of RA are termed disease-modifying antirheumatic drugs (DMARDs). Treating RA patients with DMARDs presents several advantages, including low cost, reliability, and low incidents of side effects 1. Therefore, DMARDs are often used as second-line medication in RA patients. Significant challenges arise, however, in patients that show persistent resistance to DMARDs 83. Novel strategies such as anti-TNFα blockade are therefore especially in need to treat patients not responding to DMARDs.
In 1950, Philip Hench was awarded the Nobel Prize for his application of cortisone (introduced 1949) for the treatment of RA 82. Corticosteroids were initially thought to be the cure for RA; they rapidly suppress symptoms and decrease bone erosion. However, symptoms recur following discontinuation and corticosteroids are associated with numerous profound side effects including immune, endocrine, and HPA axis suppression 82. Antimalarials including quinine and cinchonine, isolated from the cinchona tree at the same time as salicylates, were also proven effective for the treatment of RA in the early 1950s 82.
Among the DMARDs used to treat RA patients, MTX has been shown to be one of the most reliable, effective, and well-tolerated 84, 85. Since its introduction for RA in the 1980s, methotrexate (MTX) has been the gold standard treatment. Although it was initially shown to provide symptomatic relief for RA in 1951, MTX was largely used as a chemotherapeutic agent, curtailed due to the initial success of corticosteroids. MTX functions as a competitive inhibitor of dihydrofolate reductase, however, the exact mechanism of action within the context of RA is incompletely understood 82. Hypotheses include folate antagonism, adenosine signaling, generation of ROS, inhibition of angiogenesis, and alteration of cytokine profiles 86. However, MTX is still not as effective as newer treatment strategies, such as anti-TNFα, even in responding patients. Overall, the low cost and reliability of DMARDs currently confer enough advantages for them to often be considered first-line options in treating RA patients 1.
Novel or prospective treatment options
In addition to TNF-α blocking strategies, there are several other novel treatment strategies currently available or in development 1. These novel strategies include inhibitors to other pro-inflammatory cytokines such as IL-1 and IL-6, and inhibitors of more upstream inflammatory pathways, such as the Janus-activated kinase (JAK) pathway and the co-stimulatory pathways required for T-cell activation1. The IL-1 blocking agent anakinra showed lower efficacy and higher incidence of adverse effects and is therefore only recommended for patients intolerant to TNF-α blocking agents 87, 88. Co-stimulation blockers and JAK inhibitors, on the other hand, are showing promising results regarding the efficacy and adverse effects, especially in combination with MTX 89, 90. More clinical trials are needed to determine whether these newer strategies are indeed more advantageous than TNF-α blocking agents.
Standard Protocol for the Treatment of RA
Approximately 10-20% of patients do not respond to non-biologic DMARDs and are subsequently recommended for anti-TNF therapy 24. Phase I of the standard protocol for the management of RA as developed by the European League Against Rheumatism (EULAR) recommends starting the patient on MTX and short-term glucocorticoids 91. If the patient presents any contraindication for MTX, leflunomide or sulfasalazine can be used in its stead. Progression to Phase II is dependent upon symptomatic improvement; initiated if the patient does not present symptomatic improvement at 3 months and/or clinical remission at 6 months 91. Upon initiation of Phase II, if the patient does not exhibit poor prognostic factors, the DMARD is changed, or a second DMARD is added. However, if the patient does in fact present poor prognostic factors, a biological DMARD or JAK-inhibitor is added 91. Anti-TNF-α biologics fall within the former category, along with abatacept (CTLA4-Ig), IL-6 inhibitors such as tocilizumab, and rituximab, a monoclonal anti-CD20 antibody 91. Notably, methotrexate is utilized as a co-therapy in 70% of RA patients treated with anti-TNF-α biologic agents, as combination therapies have demonstrated synergistic effects 39.
Both biologic and non-biologic DMARDs may be administered orally, subcutaneously, or intravenously. Phase III treatment is initiated if symptom improvement at 3 months and/or clinical remission at 6 months is not achieved after Phase II and involves switching out the biologic DMARD or JAK-inhibitor for an agent of the same or different class 91. A 2019 meta-analysis ascertained a 24% patient remission rate (according to Disease Activity Score 28 [DAS28] remission criteria) at 24 months, indicating that despite adequate symptom management, sustained remission is rare and there is significant room for improvement insofar as unmet needs 92. Response to RA treatment, including anti-TNFs, is thought to be dependent on both genetic and environmental factors. For example, one study reported that patients who achieved DMARD-free sustained remission were rarely anti-citrullinated protein antibodies (ACPA) or rheumatoid factor-positive 93. As such, the pharmacogenomics of anti-TNF treatment response are currently under investigation using GWAS, genomic, and transcriptomic analysis methodology towards the aim of generating genetic screening methods to predict treatment response 94, 95.
Current clinical applications and future perspectives
Currently, the five TNF-α blocking agents discussed previously are FDA- and Health Canada-approved for the treatment of RA42. From a clinical perspective, TNF-α blocking agents are usually considered third-line therapy; they are prescribed in patients not responding to DMARDs such as MTX1. One of the factors precluding more widespread use of TNF-α blocking agents is its high cost as compared to MTX. Another aspect worth considering is the presence of adverse effects, especially the increased risks of infections, in patients treated with TNF-α blocking agents. It is worth pointing out that while TNF-α blocking agents are effective in limiting joint damage, they cannot reverse existing joint damage, and a substantial proportion of patients continue to demonstrate radiographic progression. Thus, TNF-α blocking agents are not cures for RA. With several new strategies showing promising results, combinational therapy has the potential of enhancing the efficacy of anti-TNF-α blockade 1,96,97. More targeted TNF-α blocking strategies have also been proposed, such as specifically targeting pro-inflammatory macrophages within the heterogeneous population implicated in RA 98,99.
Conclusion
The potent pro-inflammatory cytokine, TNF-α, plays a central role in the pathogenesis and progression of RA. Landmark in vitro, in vivo, and clinical studies conducted in the 1980s and 90s culminated in the general availability of anti-TNF-α therapies at the end of the 20th century. Currently, anti-TNFs are most commonly used in combination with MTX as part of Phase II treatment (i.e., second-line) for RA. Although patient outcomes have improved dramatically, sustained remission is rare; more research into the biology of RA, as well as the development of novel RA therapies, is warranted.
Acknowledgements
Funding Statements
The authors have no funding statements to declare.
Declaration of Competing Interests
The authors have no competing interests to declare.
Author Information
Authors and Affiliations
Department of Immunology, University of Toronto, Toronto, ON, Canada
Felicia Ceban, Katherine Jiaxi Xu
Michael G. DeGroote School of Medicine, McMaster University, Hamilton, ON, Canada
Felicia Ceban
Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada
Katherine Jiaxi Xu
Corresponding Author
Please direct correspondence to Felicia Ceban.
References
1. Abbasi, M. et al. Strategies toward rheumatoid arthritis therapy; the old and the new. J Cell Physiol 234, 10018–10031 (2019).
2. Scott, D. L., Wolfe, F. & Huizinga, T. W. J. Rheumatoid arthritis. Lancet 376, 1094–1108 (2010).
3. Rodnan, G. P. Primer on the Rheumatic Diseases: Seventh Edition. JAMA 224, 662 (1973).
4. Toussirot, É. & Wendling, D. The use of TNF-α blocking agents in rheumatoid arthritis: an overview. Expert Opinion on Pharmacotherapy 5, 581–594 (2004).
5. Feldmann, M., Brennan, F. M. & Maini, R. N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 14, 397–440 (1996).
6. McCarthy, E. F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 26, 154-158 (2006).
7. Chu, W. Tumor necrosis factor. Cancer Lett. 328, 222-225 (2013).
8. Beutler, B., Mahoney, J., Le Trang, N., Pekala, P. & Cerami, A. Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J. Exp. Med. 161, 984-995 (1985).
9. Wajant, H. & Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Frontiers in Cell and Developmental Biology 7, 91 (2019).
10. Semenzato, G. Tumour necrosis factor: a cytokine with multiple biological activities. Br. J. Cancer 61, 354-361 (1990).
11. Idriss, H. T. & Naismith, J. H. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech 50, 184–195 (2000).
12. Fernández-Ruiz, M. & Aguado, J. M. Risk of infection associated with anti-TNF-α therapy. Expert Review of Anti-infective Therapy 16, 939–956 (2018).
13. Parameswaran, N. & Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 20, 87-103 (2010).
14. Martin, C., Boisson, C., Haccoun, M., Thomachot, L. & Mege, J. Patterns of cytokine evolution (tumor necrosis factor-alpha and interleukin-6) after septic shock, hemorrhagic shock, and severe trauma. Crit. Care Med. 25 (1997).
15. Zhao, B. TNF and Bone Remodeling. Current osteoporosis reports 15, 126-134 (2017).
16. Bullock, J. et al. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med Princ Pract 27, 501–507 (2018).
17. Saklatvala, J. Tumour necrosis factor α stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 322, 547–549 (1986).
18. Arend, W. P. & Dayer, J.-M. Inhibition of the production and effects of interleukins-1 and tumor necrosis factor α in rheumatoid arthritis. Arthritis & Rheumatism 38, 151–160 (1995).
19. Firestein, G. S. Evolving concepts of rheumatoid arthritis. Nature 423, 356-361 (2003).
20. Choy, E. H. S. & Panayi, G. S. Cytokine Pathways and Joint Inflammation in Rheumatoid Arthritis. N. Engl. J. Med. 344, 907-916 (2001).
21. McInnes, I. B. & Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nature Reviews Immunology 7, 429-442 (2007).
22. Choy, E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 51, v3-v11 (2012).
23. Brennan, F. M., Chantry, D., Jackson, A. M., Maini, R. N. & Feldmann, M. Cytokine production in culture by cells isolated from the synovial membrane. J. Autoimmun. 2 Suppl, 177-186 (1989).
24. Feldmann, M. Development of anti-TNF therapy for rheumatoid arthritis. Nature Reviews Immunology 2, 364-371 (2002).
25. Kaushansky, K., Broudy, V. C., Harlan, J. M. & Adamson, J. W. Tumor necrosis factor-alpha and tumor necrosis factor-beta (lymphotoxin) stimulate the production of granulocyte-macrophage colony-stimulating factor, macrophage colony-stimulating factor, and IL-1 in vivo. J. Immunol. 141, 3410-3415 (1988).
26. Saklatvala, J., Sarsfield, S. J. & Townsend, Y. Pig interleukin 1. Purification of two immunologically different leukocyte proteins that cause cartilage resorption, lymphocyte activation, and fever. J. Exp. Med. 162, 1208-1222 (1985).
27. Butler, D. M., Maini, R. N., Feldmann, M. & Brennan, F. M. Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur. Cytokine Netw. 6, 225-230 (1995).
28. Brennan, F., Jackson, A., Chantry, D., Maini, R. & Feldmann, M. Inhibitory effect of TNFα antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. The Lancet 334, 244-247 (1989).
29. Alvaro-Gracia, J. M., Zvaifler, N. J., Brown, C. B., Kaushansky, K. & Firestein, G. S. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J. Immunol. 146, 3365-3371 (1991).
30. Williams, R. O., Feldmann, M. & Maini, R. N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. U. S. A. 89, 9784-9788 (1992).
31. Butler, D. M. et al. DBA/1 mice expressing the human TNF-alpha transgene develop a severe, erosive arthritis: characterization of the cytokine cascade and cellular composition. J. Immunol. 159, 2867-2876 (1997).
32. Keffer, J. et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025-4031 (1991).
33. Williams, R. O., Feldmann, M. & Maini, R. N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. U. S. A. 89, 9784-9788 (1992).
34. Elliott, M. J. et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor α. Arthritis & Rheumatism 36, 1681-1690 (1993).
35. Felson, D. & American College of Rheumatology Committee to Reevaluate, Improvement Criteria. A proposed revision to the ACR20: The hybrid measure of American College of Rheumatology response. Arthritis & Rheumatism 57, 193-202 (2007).
36. Feldmann, M. & Maini, S. R. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol. Rev. 223, 7-19 (2008).
37. Harding, F. A., Stickler, M. M., Razo, J. & DuBridge, R. B. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. mAbs 2, 256-265 (2010).
38. Maini, R. N. et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor α monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis & Rheumatism 41, 1552-1563 (1998).
39. Rein, P. & Mueller, R. B. Treatment with Biologicals in Rheumatoid Arthritis: An Overview. Rheumatol. Ther. 4, 247-261 (2017).
40. Lipsky, P. E. et al. Infliximab and Methotrexate in the Treatment of Rheumatoid Arthritis. N. Engl. J. Med. 343, 1594-1602 (2000).
41. Ma, X. & Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomedical Reports 1, 177–184 (2013).
42. Radner, H. & Aletaha, D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med Wochenschr 165, 3–9 (2015).
43. Maini, R. N. et al. Anti-tumour necrosis factor specific antibody (infliximab) treatment provides insights into the pathophysiology of rheumatoid arthritis. Annals of the Rheumatic Diseases 58, i56–i60 (1999).
44. Feldmann, M. & Maini, R. N. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol 19, 163–196 (2001).
45. St Clair, E. W. et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum 50, 3432–3443 (2004).
46. Lipsky, P. E. et al. Infliximab and Methotrexate in the Treatment of Rheumatoid Arthritis. N Engl J Med 343, 1594–1602 (2000).
47. Quinn, M. A. et al. Very early treatment with infliximab in addition to methotrexate in early, poor-prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve-month randomized, double-blind, placebo-controlled trial. Arthritis Rheum 52, 27–35 (2005).
48. Westhovens, R. et al. The safety of infliximab, combined with background treatments, among patients with rheumatoid arthritis and various comorbidities: a large, randomized, placebo-controlled trial. Arthritis Rheum 54, 1075–1086 (2006).
49. Vogel, W. H. Infusion reactions: diagnosis, assessment, and management. Clin J Oncol Nurs 14, E10-21 (2010).
50. Delabaye, I., De Keyser, F., & the REMITRACT study group. 74-week follow-up of safety of infliximab in patients with refractory rheumatoid arthritis. Arthritis Res Ther 12, R121 (2010).
51. van der Heijde, D. et al. Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: two-year clinical and radiographic results from the TEMPO study, a double-blind, randomized trial. Arthritis Rheum 54, 1063–1074 (2006).
52. Moreland, L. W. et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med 130, 478–486 (1999).
53. Weinblatt, M. E. et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med 340, 253–259 (1999).
54. Bathon, J. M. et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med 343, 1586–1593 (2000).
55. Klareskog, L. et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 363, 675–681 (2004).
56. Weinblatt, M. E. et al. Efficacy and safety of etanercept 50 mg twice a week in patients with rheumatoid arthritis who had a suboptimal response to etanercept 50 mg once a week: results of a multicenter, randomized, double-blind, active drug-controlled study. Arthritis Rheum 58, 1921–1930 (2008).
57. Kavanaugh, A. et al. Improvements in clinical response between 12 and 24 weeks in patients with rheumatoid arthritis on etanercept therapy with or without methotrexate. Ann Rheum Dis 67, 1444–1447 (2008).
58. Machold, K. P. & Smolen, J. S. Adalimumab – a new TNF-alpha antibody for treatment of inflammatory joint disease. Expert Opin Biol Ther 3, 351–360 (2003).
59. Breedveld, F. C. et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 54, 26–37 (2006).
60. van Schouwenburg, P. A., Rispens, T. & Wolbink, G. J. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol 9, 164–172 (2013).
61. Furst, D. E. et al. Adalimumab, a fully human anti tumor necrosis factor-alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis). J Rheumatol 30, 2563–2571 (2003).
62. van de Putte, L. B. A. et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis 63, 508–516 (2004).
63. Keystone, E. C. et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum 50, 1400–1411 (2004).
64. Weinblatt, M. E. et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 48, 35–45 (2003).
65. Takeuchi, T. et al. Effectiveness and safety of adalimumab in Japanese patients with rheumatoid arthritis: retrospective analyses of data collected during the first year of adalimumab treatment in routine clinical practice (HARMONY study). Mod Rheumatol 22, 327–338 (2012).
66. Pelechas, E., Voulgari, P. V. & Drosos, A. A. Golimumab for Rheumatoid Arthritis. J Clin Med 8, (2019).
67. Weinblatt, M. E. et al. Intravenous golimumab is effective in patients with active rheumatoid arthritis despite methotrexate therapy with responses as early as week 2: results of the phase 3, randomised, multicentre, double-blind, placebo-controlled GO-FURTHER trial. Ann Rheum Dis 72, 381–389 (2013).
68. Oldfield, V. & Plosker, G. L. Golimumab: in the treatment of rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. BioDrugs 23, 125–135 (2009).
69. Keystone, E. C. et al. Golimumab, a human antibody to tumour necrosis factor {alpha} given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis 68, 789–796 (2009).
70. Kay, J. et al. Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: A randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis & Rheumatism 58, 964–975 (2008).
71. Goel, N. & Stephens, S. Certolizumab Pegol. mAbs 2, 137–147 (2010).
72. Ruiz Garcia, V. et al. Certolizumab pegol (CDP870) for rheumatoid arthritis in adults. Cochrane Database of Systematic Reviews (2017) doi:10.1002/14651858.CD007649.pub4.
73. Radner, H. & Aletaha, D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med Wochenschr 165, 3–9 (2015).
74. Smolen, J. et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 68, 797–804 (2009).
75. Smolen, J. S. et al. Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet 388, 2763–2774 (2016).
76. Keystone, E. et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: Findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 58, 3319–3329 (2008).
77. Fleischmann, R. et al. Efficacy and safety of certolizumab pegol monotherapy every 4 weeks in patients with rheumatoid arthritis failing previous disease-modifying antirheumatic therapy: the FAST4WARD study. Ann Rheum Dis 68, 805–811 (2009).
78. Singh, J. A. et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. The Cochrane database of systematic reviews 2011, CD008794 (2011).
79. Thalayasingam, N. & Isaacs, J. D. Anti-TNF therapy. Best Practice & Research Clinical Rheumatology 25, 549-567 (2011).
80. Scheinfeld, N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J. Dermatol. Treat. 15, 280-294 (2004).
81. Ong, C. K. S., Lirk, P., Tan, C. H. & Seymour, R. A. An Evidence-Based Update on Nonsteroidal Anti-Inflammatory Drugs. Clinical Medicine & Research 5, 19–34 (2007).
82. Case, J. P. Old and new drugs used in rheumatoid arthritis: a historical perspective. Part 1: the older drugs. Am. J. Ther. 8, 123-143 (2001).
83. Bullock, J. et al. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med Princ Pract 27, 501–507 (2018).
84. Liu, D. et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol 9, 30 (2013).
85. Daien, C. I., Hua, C., Combe, B. & Landewe, R. Non-pharmacological and pharmacological interventions in patients with early arthritis: a systematic literature review informing the 2016 update of EULAR recommendations for the management of early arthritis. RMD Open 3, e000404 (2017).
86. Friedman, B. & Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine 86, 301-307 (2019).
87. Genovese, M. C. et al. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum 50, 1412–1419 (2004).
88. Cohen, S. et al. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 46, 614–624 (2002).
89. Kremer, J. M. et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum 52, 2263–2271 (2005).
90. Cohen, S. B. et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 76, 1253–1262 (2017).
91. Smolen, J. S. et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 79, 685-699 (2020).
92. Yu, C. et al. Remission rate and predictors of remission in patients with rheumatoid arthritis under treat-to-target strategy in real-world studies: a systematic review and meta-analysis. Clin. Rheumatol. 38, 727-738 (2019).
93. Ajeganova, S. & Huizinga, T. Sustained remission in rheumatoid arthritis: latest evidence and clinical considerations. Therapeutic Advances in Musculoskeletal Disease 9, 249-262 (2017).
94. Umiċeviċ Mirkov, M. et al. Genome-wide association analysis of anti-TNF drug response in patients with rheumatoid arthritis. Ann. Rheum. Dis. 72, 1375-1381 (2013).
95. Aterido, A. et al. A Combined Transcriptomic and Genomic Analysis Identifies a Gene Signature Associated With the Response to Anti-TNF Therapy in Rheumatoid Arthritis. Frontiers in immunology 10, 1459 (2019).
96. Kaneko, K. et al. Selective Inhibition of Membrane Type 1 Matrix Metalloproteinase Abrogates Progression of Experimental Inflammatory Arthritis: Synergy With Tumor Necrosis Factor Blockade. Arthritis Rheumatol 68, 521–531 (2016).
97. Aletaha, D., Funovits, J. & Smolen, J. S. Physical disability in rheumatoid arthritis is associated with cartilage damage rather than bone destruction. Ann Rheum Dis 70, 733–739 (2011).
98. Gordon, S. & Taylor, P. R. Monocyte and macrophage heterogeneity. Nat Rev Immunol 5, 953–964 (2005).
99. Udalova, I., Monaco, C., Nanchahal, J. & Feldmann, M. Anti-TNF Therapy. Microbiol Spectr 4, (2016).

